211service.com
Eerste atoomniveausimulatie van een hele batterij
Als het gaat om de ontwikkeling van de volgende generatie technologie, is het grootste knelpunt misschien wel de batterij. Ingenieurs hebben betere batterijen nodig voor elektrische voertuigen, voor energieopslag in elektriciteitsnetten en natuurlijk voor consumentenelektronica.
Deze batterijen moeten een hogere stroom leveren over meer ontlaadcycli met een grotere energiedichtheid, om maar een paar van de uitdagingen te noemen.
Het bouwen en testen van nieuwe batterijontwerpen is tijdrovend, moeilijk en duur. Het is dus handig voor elektrochemici om de werking van een batterij te simuleren voordat ze ooit hun handen vuil maken.
Dat is lastig. Niemand is in staat geweest om een volledige batterij op atomair niveau te simuleren vanwege de complexiteit van de processen die plaatsvinden en de beperkingen van de huidige modelleringstechnieken.
Vandaag verandert dat dankzij het werk van Wolf Dapp aan het Institute for Advanced Simulation en Martin Muser aan de Universiteit van Saarlandes, beide in Duitsland. Deze jongens hebben het gedrag van een hele batterij op atomaire schaal gesimuleerd. En hun simulatie reproduceert voor het eerst veel van de echte kenmerken van een batterij vanuit de eerste principes.
In de afgelopen jaren hebben computerwetenschappers aanzienlijke vooruitgang geboekt bij het simuleren van verschillende aspecten van batterijgedrag. Deze modellen werken op mesoschaal - kleiner dan de elektroden maar groter dan de moleculen. De simulaties zijn gebaseerd op experimentele gegevens om zaken als ionische en elektronische geleidbaarheid, diffusiecoëfficiënten, stroomdichtheden, elektrochemische potentialen enzovoort te modelleren.
Deze modellen hebben een serieus nadeel: ze hebben weinig voorspellende kracht als het gaat om nieuwe materialen of combinaties van materialen waarvoor geen experimentele gegevens beschikbaar zijn. Om het gedrag van nieuwe materialen te voorspellen, moeten elektrochemici batterijen modelleren op de schaal van atomen en moleculen.
Dat is lastig, want de technieken die computerwetenschappers gebruiken om het gedrag van atomen en moleculen te simuleren, zijn niet geschikt voor batterijen. Deze simulaties zijn ontworpen voor systemen die in evenwicht zijn of er dichtbij zijn. Ze werken door het chemische potentieel gelijk te maken of de energie van het systeem te minimaliseren. Het verschil in chemische potentiaal tussen twee elektroden is echter precies wat het ladingstransport in een batterij aandrijft, zeggen Dapp en Muser.
Dus om een batterij als geheel te modelleren, moet het computermodel bij elke stap in de berekening rekening houden met elke verandering in energie of chemisch potentieel. Dit is precies wat Dapp en Muser hebben gedaan. In hun model is lading een variabele die in elke fase van de berekening kan worden uitgewisseld tussen atomen en tussen bindingen.
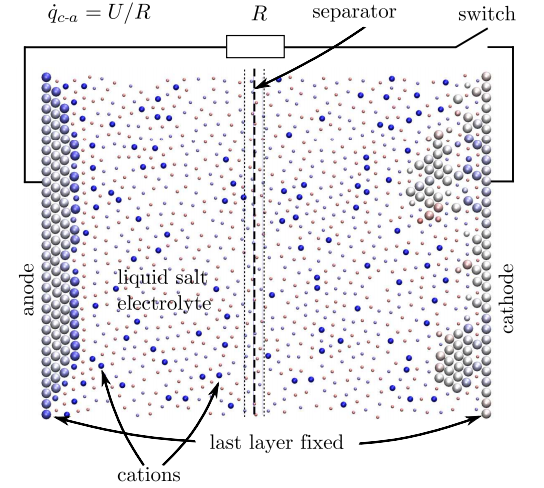
De resulterende simulaties zijn klein maar indrukwekkend. Hun nanobatterij bestaat uit 358 atomen, waarvan 118 de elektroden vormen. De kathode is aanvankelijk bedekt met een laag van 20 atomen met 39 positief geïoniseerde atomen opgelost in de elektrolyt.
De berekening verloopt dan in stappen waarin atomen kunnen bewegen en lading kunnen uitwisselen terwijl het systeem evolueert. De hele simulatie bestaat uit zo'n 10 miljoen van deze stappen.
De resultaten zijn opmerkelijk omdat ze de algemene ontlaadcurven van echte macroscopische batterijen reproduceren. Een lagere bedrijfstemperatuur vermindert bijvoorbeeld de effectieve capaciteit van de gesimuleerde batterij. En het allerbelangrijkste: de simulatie reproduceert de manier waarop gewone batterijen verslijten. Bij het opladen nemen de prestaties van de batterij iets af en verandert de morfologie van het elektrode-oppervlak tijdens het gebruik van de batterij, zeggen Dapp en Muser.
Deze jongens zeggen dat het werk in dit stadium slechts een proof-of-principle-model is en dat er verschillende manieren zijn waarop het in de toekomst kan worden verbeterd. Ze modelleren bijvoorbeeld de elektrolyt met deeltjes die een vaste lading hebben en deze dus niet kunnen uitwisselen.
Dat is niet hoe elektrolyten werken in echte batterijen en dit is dus een belangrijke tekortkoming van de nieuwe methode. Maar Dapp en Muser zijn van plan dit te corrigeren. Deze idealisering zal in toekomstig werk worden opgegeven, zeggen ze.
Al met al lijkt dit belangrijk werk te zijn. Dit soort model zou de voorspellende kracht van batterijsimulaties drastisch kunnen verbeteren en daardoor elektrochemici veel tijd, energie en geld kunnen besparen voordat ze met gedetailleerde experimenten beginnen.
Het eindresultaat zou betere batterijen moeten zijn, maar er is nog een lange weg te gaan.
Referentie: arxiv.org/abs/1308.3424 : Redoxreacties met empirische mogelijkheden: simulaties van atomaire ontlading van batterijen