211service.com
Shannon's wiskundige communicatietheorie toegepast op DNA-sequencing
Een van de grote onbezongen helden van de 20e-eeuwse wetenschap is Claude Shannon, een ingenieur bij de beroemde Bell Laboratories tijdens zijn hoogtijdagen in het midden van de 20e eeuw. Shannons meest blijvende bijdrage aan de wetenschap is de informatietheorie, die ten grondslag ligt aan alle digitale communicatie.
In een beroemd artikel uit het einde van de jaren veertig schetste Shannon het fundamentele probleem van communicatie: op het ene punt in de ruimte een bericht reproduceren dat op een ander punt is gecreëerd. Het bericht wordt eerst op de een of andere manier gecodeerd, verzonden en vervolgens gedecodeerd.
Shannon's toonde aan dat een bericht altijd met willekeurige precisie op een ander punt in de ruimte kan worden gereproduceerd, op voorwaarde dat de ruis onder een bepaald drempelniveau blijft. Hij ging verder met berekenen hoeveel informatie op deze manier kon worden verzonden, een eigenschap die bekend staat als de capaciteit van dit informatiekanaal.
Shannons ideeën zijn met veel succes op grote schaal toegepast op alle vormen van informatieoverdracht. Een bijzonder interessante weg was de toepassing van informatietheorie op biologie - het idee dat het leven zelf de overdracht van informatie van de ene generatie naar de volgende is.
Dat soort denken is aan de gang, revolutionair en staat nog in de kinderschoenen. Er komt veel.
Vandaag kijken we naar een interessant gevolg op het gebied van biologische informatieoverdracht. Abolfazl Motahari en vrienden van de University of California, Berkeley, gebruiken Shannons benadering om te onderzoeken hoe snel informatie uit DNA kan worden gehaald met behulp van shotgun-sequencing.
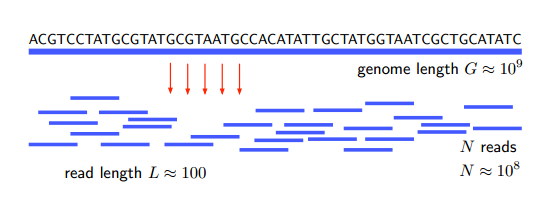
Het probleem hier is om de volgorde van nucleotiden (A, G, C en T) in een genoom te bepalen. Dat is tijdrovend omdat genomen vaak lang zijn - het menselijk genoom bestaat bijvoorbeeld uit zo'n 3 miljard nucleotiden of basenparen. Dit zou een eeuwigheid duren om in serie te sequensen.
Dus de shotgun-benadering houdt in dat het genoom in willekeurige stukjes wordt geknipt, bestaande uit 100 tot 1.000 basenparen, en deze parallel wordt gerangschikt. De informatie wordt dan weer aan elkaar gelijmd in silico door een zogenaamd reassembly-algoritme.
Natuurlijk is er geen manier om te weten hoe je de informatie opnieuw kunt samenstellen uit een enkele lezing van het genoom. Dus in de shotgun-aanpak wordt dit proces vele malen herhaald. Omdat elke read het genoom op een andere manier verdeelt, overlappen stukken onvermijdelijk met segmenten van een vorige run. Deze overlapgebieden maken het mogelijk om het hele genoom weer in elkaar te zetten, als een legpuzzel.
Dat ruikt naar een klassiek probleem van de informatietheorie, en inderdaad hebben verschillende mensen op deze manier nagedacht. Motahari en co gaan echter een stap verder door het min of meer precies te herformuleren als een analoog van Shannons beroemde benadering.
Ze zeggen dat het probleem van genoomsequencing in wezen bestaat uit het reproduceren van een bericht geschreven in DNA, in een digitaal elektronisch formaat. Bij deze benadering bevindt het originele bericht zich in het DNA, wordt het gecodeerd voor verzending door het leesproces en wordt het vervolgens gedecodeerd door een hermontage-algoritme om een elektronische versie te produceren.
Wat ze bewijzen, is dat er een kanaalcapaciteit is die een maximale snelheid van informatiestroom definieert tijdens het sequencingproces. Het geeft het maximale aantal DNA-basenparen dat per lezing kan worden opgelost, door elk assemblage-algoritme, zonder rekening te houden met computationele beperkingen, zeggen ze.
Dat is een significant resultaat voor iedereen die geïnteresseerd is in het sequencen van genomen. Een belangrijke vraag is hoe snel een bepaalde sequencing-technologie zijn werk kan doen en of het sneller of langzamer is dan andere benaderingen.
Dat is op dit moment niet mogelijk, omdat veel van de algoritmen die voor assemblage worden gebruikt, zijn ontworpen voor specifieke technologieën en benaderingen van lezen. Motohari en co zeggen dat er bijvoorbeeld minstens 20 verschillende hermontage-algoritmen zijn. Dit maakt het moeilijk om verschillende algoritmen met elkaar te vergelijken, zeggen ze.
Bijgevolg weet niemand echt wat het snelst is of zelfs wat het potentieel heeft om het snelst te zijn.
Het nieuwe werk brengt daar verandering in. Voor het eerst zou het mogelijk moeten zijn om te werken hoe dicht een bepaalde sequencing-technologie bij de theoretische limiet komt.
Dat zou wel eens een dood hout uit dit gebied kunnen verdringen en een periode van snelle innovatie in sequencing-technologie kunnen stimuleren.
Referentie: arxiv.org/abs/1203.6233 : Informatietheorie van DNA-sequencing